Can’t Remember? Is Your Protein Bent?
READERS SUMMARY:
1. Alzheimer’s disease is due to lack of electrons which causes compliant design changes in protein phosphatase!
2. Why are the incidence and prevalence growing for AD?
3. What is a proteopathy?
4. Why is protein folding so critical to so many diseases?
5. What kind of nutrition guidelines do I follow to avoid AD doc?
Many of you had questions after my first Alzheimer’s Disease post (AD) but few seem to want me to call the cause of it. The truth is no one is willing to make the leap of faith just yet, but the data that we have found over the last ten years has given us a pretty good idea of what causes it to occur. I am not going to be so kind. AD is the major neolithic disease coming from People 0-40 years old today. No Neolithic disease we have seen be born in the last 125 years will have the impact on our society that AD will have. Humans evolved from other hominids because of their brain and how they rely on social learning. AD completely destroys that beautiful adaptation. Many younger people think that AD is a disease of the elderly brain. I have some bad news for you. It is not. It is a neolithic disease whose onset is tied some critical decisions about the fuel choice you make for your brain. My prediction is that the next fifty years we will see AD begin to afflict many thousands of people below the age of fifty and it won’t be because they have a problem with their APo E 4 allele.
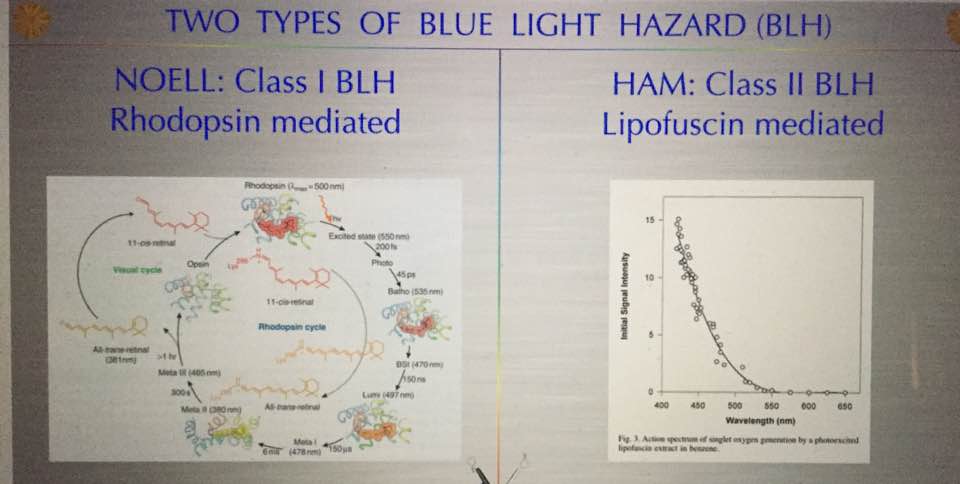
Alzheimer’s is a proteopathy that is affected by incident light frequencies via the retina. A proteopathy is a disease caused by altered protein size or shape. For example, yellow fat disease known as lipofuscinosis is one example of proteinopathy besides AD. Lipofuscinosis is seen in the blue light hazard we see in the retina. The shape and size change is mediated by a lack of electrons (DHA in retina) in certain proteins in the brain which controls metabolism and energy utilization. A proteopathy is a disease that at its core has misfolded proteins in its etiology. It appears that AD is caused by several key factors that all seem to show up in unison together. One is an extreme lack of lipids (cholesterol) in the brain. The lipids missing tend to be omega 3 fat variety. Cholesterol and the phospholipids are the others that are missing in action. But even more interesting it appears the brain needs between 25-35% of omega 3 fats called DHA to function optimally. There also needs to be comorbid neuronal intracellular inflammation (protonopathy) present. In this situation, we also see up-regulation of the IGF-1 pathways and dysregulation of the protein signaling that leads to faulty protein folding in the AD brain. We used to think the Tau protein that caused the neurofibrillary tangle were the result of the disease but it is becoming clear that the real issue is that these are remnants of faulty protein folding and non-functional neurons result. The neurons with these misfolded proteins are toxic and need to be replaced so they undergo apoptosis instead of autophagy and the brain’s stem cell supply in the temporal, parietal and frontal lobes become exhausted. This begs a major question then? Why is protein folding so important and what controls it? Is this tied to changes in electrons and protons in cells due to the light environment?
Well, this is where we have to hit some hardcore biochemistry/biophysics now so rub your head now! Protein kinases are enzymes that donate phosphate groups from energy sources like ATP to other proteins. This process, called phosphorylation, is what controls protein folding. The addition or subtraction of electrons to these proteins is what controls this process. This process is called phosphorylation. Most of you know ATP is the source of phosphatase biology. Both cell motility and cAMP-induced cAMP production are regulated by phosphatidylinositol 3-kinase (PI3K)-dependent pathways and this is linked to the pulses of cAMP a tissue can generate.
This biologic step is reversible as well so it can go in either direction. It allows for flexibility in the system during growth and generating metabolic flexibility. The interesting part of this process is that each addition or subtraction of a phosphate group causes a conformational change in the structure of the protein. This means its size or shape changes. When its shape changes it also alters its biologic function as well. This changes the way the protein interacts with other proteins or how signaling occurs in the cell. Most cellular receptors use phosphorylation in order to record the chemical signals. This occurs with all hormone receptors for example. We know in AD there is a big association with diabetes because of glucose metabolism and cAMP second messenger system.
PIP3 also recruits and activates a number of PH (phosphate) domain proteins, including protein kinase B (Akt) and cytosolic regulator of adenylyl cyclase (CRAC). Akt regulates cell motility by regulating cytoskeleton remodeling, and CRAC is required for the activation of adenylyl cyclase (AC), which then catalyzes cAMP production (1, 2, 3). Cells lacking PI3K1 and PI3K2 have defective responses to chemoattractant (4). In contrast, cells lacking phosphatase and tensin homolog (PTEN), a phosphatase that dephosphorylates PIP3, have extended responses to cAMP stimulation (5, 6). This process is broken down in the AD retina and brain to cause a circadian mismatch and ruining the SCN function. This is why AD is often associated with daytime sleepiness, poor sleep at night, and cognitve issue decades before frank AD is diagnosed. The changes in the eye and brain are caused by changes in protein size and shape. This is why retinal changes can be seen in diabetics and with enough mitochondrial damage they begin to show up in the frontal temporal lobes of the brain.

Diabetes increases the risk of developing Alzheimer’s disease by 147 times according to a recent Science Daily post! Since you all know we have anywhere between 60-150 million Americans with diabetes or pre-diabetes you can see the “table is set” for AD to completely explode in the next several generations. Some people refer to AD as type three diabetes today. The reason is that in diabetes there are high levels of insulin present and increased levels of IGF-1 (insulin growth factor 1) and Epidermal growth factor (EGF) signaling that occurs. These two receptors also rely on phosphorylation (en.wikipedia.org/wiki/Phosphorylation) for their own signal transduction as well. It appears in AD when insulin levels are high or IGF-1 levels are high, so is HS-CRP. Remember this is an inflammatory marker made in the liver. When these things occur in concert we begin to see the protein kinases of the neurons constantly kept in the “on” position. This means that they are constantly changing the signal transduction of the receptor sites by inducing folding changes in the IGF-1 and EGF receptors. This process is called hyperphosphorylation. This creates a signal to noise in concert with constant elevated inflammation, insulin levels, leptin levels, and cortisol levels. This all induces more kinase activation that eventually causes signaling changes that lead to the formation of tau proteins and tangles that characterizes AD development. All these actions cause a loss of electrons because of the poor signaling.
Once these proteins are made in neurons they induce chemical changes within the neuron to activate apoptosis and the cell commits suicide reducing the neuronal pool and eventually affecting cognition when neuron numbers reach critically low levels in our brains. Hyperphosphorylation of the tau protein can result in the self-assembly of tangles of paired helical filaments and straight filaments, which are likely involved in the pathogenesis of Alzheimer’s disease and other tauopathies. Some people believe that this is not a pathologic finding and represents the neuron trying to heal itself and overcome the misfolding. When the neuron dies and leaves behind the misfolded insoluble Tau protein. Normal Tau proteins are very soluble and tend to be found only in neurons. Tau protein’s normal job in the brain is to stabilize microtubules. Microtubules are the foundational building block for consciousness to have evolved in the human brain. In fact, it is now becoming clear that quantum biologic effects between microtubules in the brain are why we sleep, how we go are able to go “asleep” via anesthetic gases and how we dream. This process is governed by something called gamma coherence. I will blog about that in the future. Several neurodegenerative and other diseases are believed to result from the accumulation of amyloid fibrils formed by misfolded proteins as well. Parkinson’s disease, Huntington’s disease, Mad Cow disease, Pick’s disease is just a few you may have heard spoken of.
I told you that protein kinases are the “turn on” switch that causes misfolding protein in AD brains but what turns “off the switch”? The off button is performed by a protein phosphatase. PTEN is one of those phosphatases and has been shown to also have disordered signaling in neurodegeneration. If you can not turn off the protein kinases you wind up with the same diseases that are caused by hyper-phosphorylation. This data is from my last cite in the footnotes. Most pharma research has been focused on the kinase “turn on” switches and very little work has been done on the “off switches“. PTEN, mentioned above, is the off switch for this circuit. In vivo, most phosphatases also double as tumor suppressor genes which further interests me in exploring this area of biology in AD. The reason I have focused on PTEN, is because, after P53 (the guardian of our genome), it is the most mutated gene in human cancers and in mitochondrial diseases associated with cognitive defects. This means it is important and worthy of our attention in the QUILT treatise. It also has been shown to directly protect P53 in many cancer research studies. We also know that lower cholesterol levels are correlated with cancer generation while also being seen in the developing brains of AD patients. That connection is not lost on me and I think there is maybe more than meets the eye to this story. PTEN is linked with obesity and T2D as well. PTEN mutations increase the risk of obesity and cancers but they tend to decrease the risk of type 2 diabetes via enhanced insulin sensitivity because of how cAMP is pulsed in cells to control protein bending. It appears the size and change of the PTEN mutation are what is critical in the development of AD, obesity, and T2D. PTEN is an important processing molecule of signals in cells because it has roles in both cellular growth and metabolic signaling. Germline PTEN mutations cause a cancer-predisposition Cowden’s syndrome. Understanding its biology will provide an opportunity to study the effect of PTEN haploinsufficiency in humans.
I think the phosphoproteomic studies being done in epigenomic cancer research may give us some huge clues in the direct mechanism of how AD develops. I believe these will be very similar to the prion diseases. These epigenomic wide analyses are beginning to show huge interconnected system-wide connections to changes in protein kinase activation and phosphatase deactivation by the addition and subtraction of electrons. Many of our older theories on AD are being blown up by the new data coming out of new academic research. The lay public need to be kept informed of these new findings. I personally think AD may help us uncover many other scientific facts that hopefully will continue to destroy the lipid hypothesis and the heart lipid hypothesis. These two theories have caused more morbidity and mortality than cures in my opinion.
We need to maintain an open mind in neurodegenerative disease causation, but it is clear what correlates to this disease……..
1. Heavy dietary intake of carbohydrates out of season.
2. An excess of Omega 6 Polyunsaturated fats in cell membranes affects protons in cells.
3. Simultaneous deficiency of dietary Omega 3 fats in cell membranes decreasing electrons in lipoproteins.
4. Trauma that is repetitive that increases injury and lowers cortisol and melatonin levels which deplete DHA levels in the retina and brain
5. Constant pressures to switch on epigenetic programming to cause misfolding of our proteins by withdrawing electrons and causing a protonopathy in the metabolic machinery of cells.
Non-functional proteins that uncouple mitochondria from metabolism and lead to early apoptosis and this drains our neuronal stem cell pool over a lifetime to lead to a loss of neurons and loss of brain mass.
This is why we see the loss of neurons on CT’s scans and MRI’s in my clinic in people with blue light toxicity. We also can see retinal changes way before the brain shrinks if we look.
AD is preventable and avoidable if you choose to avoid circadian mismatches and malnutrition laid out above and make seafood and seasonally saturated fat a big part of your environment! Malnutrition is not just caused by a lack of some food or mineral. The most common cause is eating things outside of the proper solar electromagnetic footprint of seasons. Each season has a power density that programs electrons and protons for proper mitochondrial function.
CITES
1. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (June 2001). “Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments”. Proc. Natl. Acad. Sci. U.S.A. 98 (12): 6923-8. doi:10.1073/pnas.121119298. PMC 34454. PMID 11381127.
2. Dennis J. Selkoe (2003). “Folding proteins in fatal ways”. Nature 426 (6968): 900-904. doi:10.1038/nature02264. PMID 14685251.
3. Alberts, Bruce, Dennis Bray, Karen Hopkin, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter. “Protein Structure and Function.” Essential Cell Biology. Edition 3. New York: Garland Science, Taylor and Francis Group, LLC, 2010. Pg 120-170.
4. http://evolutionarypsychiatry.blogspot.com/2011_06_01_archive.html
5. http://evolutionarypsychiatry.blogspot.com/2011/06/nutrition-and-alzheimers-disease.html
6. J Amer Geriatric Soc. 2010 Mar;58(3):487-92. Metabolic syndrome and risk of dementia in older adults.
7. http://diss.kib.ki.se/2005/91-7140-514-3/thesis.pdf
8. http://online.wsj.com/article/SB10001424052702303661904576452243498496516.html?mod=rss_Health
9.http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(13)60453-5/abstract