

READERS SUMMARY:
1. Alzheimer’s disease is due to lack of electrons which causes compliant design changes in protein phosphatase!
2. Why are the incidence and prevalence growing for AD?
3. What is a proteopathy?
4. Why is protein folding so critical to so many diseases?
5. What kind of nutrition guidelines do I follow to avoid AD doc?
Many of you had questions after my first Alzheimer’s Disease post (AD) but few seem to want me to call the cause of it. The truth is no one is willing to make the leap of faith just yet, but the data that we have found over the last ten years has given us a pretty good idea of what causes it to occur. I am not going to be so kind. AD is the major neolithic disease coming from People 0-40 years old today. No Neolithic disease we have seen be born in the last 125 years will have the impact on our society that AD will have. Humans evolved from other hominids because of their brain and how they rely on social learning. AD completely destroys that beautiful adaptation. Many younger people think that AD is a disease of the elderly brain. I have some bad news for you. It is not. It is a neolithic disease whose onset is tied some critical decisions about the fuel choice you make for your brain. My prediction is that the next fifty years we will see AD begin to afflict many thousands of people below the age of fifty and it won’t be because they have a problem with their APo E 4 allele.
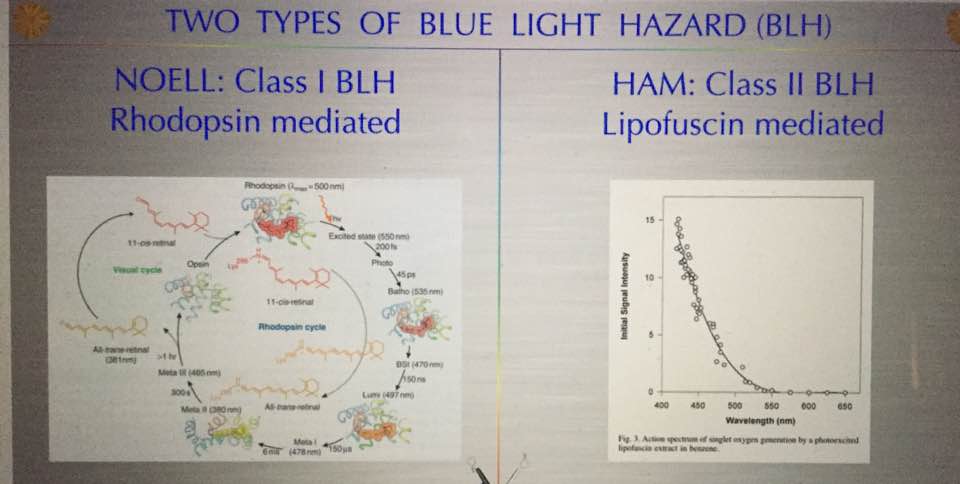
Alzheimer’s is a proteopathy that is affected by incident light frequencies via the retina. A proteopathy is a disease caused by altered protein size or shape. For example, yellow fat disease known as lipofuscinosis is one example of proteinopathy besides AD. Lipofuscinosis is seen in the blue light hazard we see in the retina. The shape and size change is mediated by a lack of electrons (DHA in retina) in certain proteins in the brain which controls metabolism and energy utilization. A proteopathy is a disease that at its core has misfolded proteins in its etiology. It appears that AD is caused by several key factors that all seem to show up in unison together. One is an extreme lack of lipids (cholesterol) in the brain. The lipids missing tend to be omega 3 fat variety. Cholesterol and the phospholipids are the others that are missing in action. But even more interesting it appears the brain needs between 25-35% of omega 3 fats called DHA to function optimally. There also needs to be comorbid neuronal intracellular inflammation (protonopathy) present. In this situation, we also see up-regulation of the IGF-1 pathways and dysregulation of the protein signaling that leads to faulty protein folding in the AD brain. We used to think the Tau protein that caused the neurofibrillary tangle were the result of the disease but it is becoming clear that the real issue is that these are remnants of faulty protein folding and non-functional neurons result. The neurons with these misfolded proteins are toxic and need to be replaced so they undergo apoptosis instead of autophagy and the brain’s stem cell supply in the temporal, parietal and frontal lobes become exhausted. This begs a major question then? Why is protein folding so important and what controls it? Is this tied to changes in electrons and protons in cells due to the light environment?
Well, this is where we have to hit some hardcore biochemistry/biophysics now so rub your head now! Protein kinases are enzymes that donate phosphate groups from energy sources like ATP to other proteins. This process, called phosphorylation, is what controls protein folding. The addition or subtraction of electrons to these proteins is what controls this process. This process is called phosphorylation. Most of you know ATP is the source of phosphatase biology. Both cell motility and cAMP-induced cAMP production are regulated by phosphatidylinositol 3-kinase (PI3K)-dependent pathways and this is linked to the pulses of cAMP a tissue can generate.
This biologic step is reversible as well so it can go in either direction. It allows for flexibility in the system during growth and generating metabolic flexibility. The interesting part of this process is that each addition or subtraction of a phosphate group causes a conformational change in the structure of the protein. This means its size or shape changes. When its shape changes it also alters its biologic function as well. This changes the way the protein interacts with other proteins or how signaling occurs in the cell. Most cellular receptors use phosphorylation in order to record the chemical signals. This occurs with all hormone receptors for example. We know in AD there is a big association with diabetes because of glucose metabolism and cAMP second messenger system.
PIP3 also recruits and activates a number of PH (phosphate) domain proteins, including protein kinase B (Akt) and cytosolic regulator of adenylyl cyclase (CRAC). Akt regulates cell motility by regulating cytoskeleton remodeling, and CRAC is required for the activation of adenylyl cyclase (AC), which then catalyzes cAMP production (1, 2, 3). Cells lacking PI3K1 and PI3K2 have defective responses to chemoattractant (4). In contrast, cells lacking phosphatase and tensin homolog (PTEN), a phosphatase that dephosphorylates PIP3, have extended responses to cAMP stimulation (5, 6). This process is broken down in the AD retina and brain to cause a circadian mismatch and ruining the SCN function. This is why AD is often associated with daytime sleepiness, poor sleep at night, and cognitve issue decades before frank AD is diagnosed. The changes in the eye and brain are caused by changes in protein size and shape. This is why retinal changes can be seen in diabetics and with enough mitochondrial damage they begin to show up in the frontal temporal lobes of the brain.

Diabetes increases the risk of developing Alzheimer’s disease by 147 times according to a recent Science Daily post! Since you all know we have anywhere between 60-150 million Americans with diabetes or pre-diabetes you can see the “table is set” for AD to completely explode in the next several generations. Some people refer to AD as type three diabetes today. The reason is that in diabetes there are high levels of insulin present and increased levels of IGF-1 (insulin growth factor 1) and Epidermal growth factor (EGF) signaling that occurs. These two receptors also rely on phosphorylation (en.wikipedia.org/wiki/Phosphorylation) for their own signal transduction as well. It appears in AD when insulin levels are high or IGF-1 levels are high, so is HS-CRP. Remember this is an inflammatory marker made in the liver. When these things occur in concert we begin to see the protein kinases of the neurons constantly kept in the “on” position. This means that they are constantly changing the signal transduction of the receptor sites by inducing folding changes in the IGF-1 and EGF receptors. This process is called hyperphosphorylation. This creates a signal to noise in concert with constant elevated inflammation, insulin levels, leptin levels, and cortisol levels. This all induces more kinase activation that eventually causes signaling changes that lead to the formation of tau proteins and tangles that characterizes AD development. All these actions cause a loss of electrons because of the poor signaling.
Once these proteins are made in neurons they induce chemical changes within the neuron to activate apoptosis and the cell commits suicide reducing the neuronal pool and eventually affecting cognition when neuron numbers reach critically low levels in our brains. Hyperphosphorylation of the tau protein can result in the self-assembly of tangles of paired helical filaments and straight filaments, which are likely involved in the pathogenesis of Alzheimer’s disease and other tauopathies. Some people believe that this is not a pathologic finding and represents the neuron trying to heal itself and overcome the misfolding. When the neuron dies and leaves behind the misfolded insoluble Tau protein. Normal Tau proteins are very soluble and tend to be found only in neurons. Tau protein’s normal job in the brain is to stabilize microtubules. Microtubules are the foundational building block for consciousness to have evolved in the human brain. In fact, it is now becoming clear that quantum biologic effects between microtubules in the brain are why we sleep, how we go are able to go “asleep” via anesthetic gases and how we dream. This process is governed by something called gamma coherence. I will blog about that in the future. Several neurodegenerative and other diseases are believed to result from the accumulation of amyloid fibrils formed by misfolded proteins as well. Parkinson’s disease, Huntington’s disease, Mad Cow disease, Pick’s disease is just a few you may have heard spoken of.
I told you that protein kinases are the “turn on” switch that causes misfolding protein in AD brains but what turns “off the switch”? The off button is performed by a protein phosphatase. PTEN is one of those phosphatases and has been shown to also have disordered signaling in neurodegeneration. If you can not turn off the protein kinases you wind up with the same diseases that are caused by hyper-phosphorylation. This data is from my last cite in the footnotes. Most pharma research has been focused on the kinase “turn on” switches and very little work has been done on the “off switches“. PTEN, mentioned above, is the off switch for this circuit. In vivo, most phosphatases also double as tumor suppressor genes which further interests me in exploring this area of biology in AD. The reason I have focused on PTEN, is because, after P53 (the guardian of our genome), it is the most mutated gene in human cancers and in mitochondrial diseases associated with cognitive defects. This means it is important and worthy of our attention in the QUILT treatise. It also has been shown to directly protect P53 in many cancer research studies. We also know that lower cholesterol levels are correlated with cancer generation while also being seen in the developing brains of AD patients. That connection is not lost on me and I think there is maybe more than meets the eye to this story. PTEN is linked with obesity and T2D as well. PTEN mutations increase the risk of obesity and cancers but they tend to decrease the risk of type 2 diabetes via enhanced insulin sensitivity because of how cAMP is pulsed in cells to control protein bending. It appears the size and change of the PTEN mutation are what is critical in the development of AD, obesity, and T2D. PTEN is an important processing molecule of signals in cells because it has roles in both cellular growth and metabolic signaling. Germline PTEN mutations cause a cancer-predisposition Cowden’s syndrome. Understanding its biology will provide an opportunity to study the effect of PTEN haploinsufficiency in humans.
I think the phosphoproteomic studies being done in epigenomic cancer research may give us some huge clues in the direct mechanism of how AD develops. I believe these will be very similar to the prion diseases. These epigenomic wide analyses are beginning to show huge interconnected system-wide connections to changes in protein kinase activation and phosphatase deactivation by the addition and subtraction of electrons. Many of our older theories on AD are being blown up by the new data coming out of new academic research. The lay public need to be kept informed of these new findings. I personally think AD may help us uncover many other scientific facts that hopefully will continue to destroy the lipid hypothesis and the heart lipid hypothesis. These two theories have caused more morbidity and mortality than cures in my opinion.
We need to maintain an open mind in neurodegenerative disease causation, but it is clear what correlates to this disease……..
1. Heavy dietary intake of carbohydrates out of season.
2. An excess of Omega 6 Polyunsaturated fats in cell membranes affects protons in cells.
3. Simultaneous deficiency of dietary Omega 3 fats in cell membranes decreasing electrons in lipoproteins.
4. Trauma that is repetitive that increases injury and lowers cortisol and melatonin levels which deplete DHA levels in the retina and brain
5. Constant pressures to switch on epigenetic programming to cause misfolding of our proteins by withdrawing electrons and causing a protonopathy in the metabolic machinery of cells.
Non-functional proteins that uncouple mitochondria from metabolism and lead to early apoptosis and this drains our neuronal stem cell pool over a lifetime to lead to a loss of neurons and loss of brain mass.
This is why we see the loss of neurons on CT’s scans and MRI’s in my clinic in people with blue light toxicity. We also can see retinal changes way before the brain shrinks if we look.
AD is preventable and avoidable if you choose to avoid circadian mismatches and malnutrition laid out above and make seafood and seasonally saturated fat a big part of your environment! Malnutrition is not just caused by a lack of some food or mineral. The most common cause is eating things outside of the proper solar electromagnetic footprint of seasons. Each season has a power density that programs electrons and protons for proper mitochondrial function.
CITES
1. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (June 2001). “Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments”. Proc. Natl. Acad. Sci. U.S.A. 98 (12): 6923-8. doi:10.1073/pnas.121119298. PMC 34454. PMID 11381127.
2. Dennis J. Selkoe (2003). “Folding proteins in fatal ways”. Nature 426 (6968): 900-904. doi:10.1038/nature02264. PMID 14685251.
3. Alberts, Bruce, Dennis Bray, Karen Hopkin, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter. “Protein Structure and Function.” Essential Cell Biology. Edition 3. New York: Garland Science, Taylor and Francis Group, LLC, 2010. Pg 120-170.
4. http://evolutionarypsychiatry.blogspot.com/2011_06_01_archive.html
5. http://evolutionarypsychiatry.blogspot.com/2011/06/nutrition-and-alzheimers-disease.html
6. J Amer Geriatric Soc. 2010 Mar;58(3):487-92. Metabolic syndrome and risk of dementia in older adults.
7. http://diss.kib.ki.se/2005/91-7140-514-3/thesis.pdf
8. http://online.wsj.com/article/SB10001424052702303661904576452243498496516.html?mod=rss_Health
9.http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(13)60453-5/abstract
I totally agree with you. What I like about your writing is that you did not fill it with unnecessary fluff. Most of the times, less is more and your opinion on this matter definitely highlights that. Clear and concise, very informative, and I will definitely be back for more. Thanks for the useful info!
Jack,
Off topic but I corresponded with you via email earlier this month. I am a young 22 year old male with testosterone levels averaging around 300-400 in the afternoon. I am pretty sure my decresed testosterone has alot to do with my chronic melatotin supplementation for 2 years, as well as a bit of undereating and overtraining. I have recently stopped this and noticed increases in T via morning erections, better libdo, but I am wondering if this can still be fully recovered. From my understanding, melatonin decreases GnRH and thus testosterone… can this be reversed? How long can it take? I have read up to 6 months for some…
@Bill. I did get your email and yes it can be reversed. It takes time for the brain to recalibrate but generally this should reverse in 3-6 months. If you want it to happen faster you can "facilitate" it by turning off all your lights as soon as the sun sets and blacking out your sleeping area. I also recommend listening to quiet relaxing music while you dark adapt or advocate activities that release oxytocin. (orgasm or nipple massage) I also recommend using L-theanine to help as well. Believe it or not men can get defects in progesterone metabolism with prolonged melatonin use and this will also effect their ability to sleep as well as their libido. Not sure this has anything to do with todays post but I hope this helps you.
J Androl. 2002 Jul-Aug;23(4):572-8.
Melatonin administration alters semen quality in healthy men.
Luboshitzky R, Shen-Orr Z, Nave R, Lavi S, Lavie P.
Department of Endocrinology, Haemek Medical Center, Afula, Israel. luboshitzky_r@clalit.org.il
The role of melatonin in the regulation of reproduction in humans is unknown. We conducted a 6-month, double-blind, crossover study of a daily treatment dose of 3 mg melatonin or placebo given orally at 1700 hours in 8 healthy men. Semen quality (concentration, motility, and morphology), serum and seminal plasma 17-beta-estradiol (E(2)), testosterone, melatonin, and serum gonadotropin levels were determined every 3 months throughout the study. In 6 men, there was no change in semen quality or in serum and seminal plasma hormone levels during the study period. In 2 men, during the melatonin treatment period, sperm concentration decreased to 3 x 10(6)/mL and 12 x 10(6)/mL, and motility declined to 32% and 30%. These coincided with a decline in seminal plasma and serum E(2) levels and with an increase in testosterone:E(2) ratios. Six months after the cessation of melatonin, sperm concentration and motility were normal in 1 man but remained abnormal in the other one with a still elevated testosterone:E(2) ratio. Serum gonadotropin levels were unchanged during the study in all 8 men. Our preliminary observations suggest that long-term melatonin administration is associated with decreased semen quality in a number of healthy men, probably through the inhibition of aromatase at the testicular level.
PMID: 12065466 [PubMed – indexed for MEDLINE]
My son was just told his triglycerides are over 500 and that his liver is in trouble. He has fat deposits, bumps that are visible. He was told absolutely nothing with fat. He is 25. He never was fat, but in the last year, the weight has come. Also, his infant son is having some severe health problems. Any ideas how to help this?
Yes but the advice he is going to get and what I would do are completely opposite. If I were him, I would go on a strict paleolithic diet and consume alot of offal and omega three eggs. He needs a ton of choline rich foods to get the fat out of his liver. I would also be in very high dose marine fish oil of Rx grader quality and make sure I was in hormonal balance with leptin and cortisol.
Thank you a bunch for sharing this with all folks
your welcome
Im grateful for the blog post. Keep writing.
Helps alot! Thanks Doc!
Wow, incredible weblog.
Is it possible that the higher mortality rate in those with lower cholesterol has something to do with the use of statins?
Not well established. But I will tell you the one thing I am paying close attention to is the current CETP trials. These drugs lowed LDL to unreal low levels. And Framingham's data predicts that if we artificially lower LDL's to low levels they will die and die early of cancer. This first trial on a CETP drug was an epic failure for the drug companies. Now the cardiologists have made every excuse in the book and believe that studying the subgroups for good effects is now worth it. Well pay attention folks. I bet the CETP trials will be the beginning of the end for Ancel Key's lipid hypothesis. It is critical to get rid of it because I see no tipping point occurring for health until paleolithic eating is routinely the advice given to patients. Since conventional wisdom says fat causes heart disease the lipid hypothesis must be destroyed. The data already is pretty clear but the house of cards is supported by big pharma and docs on the payrolls. My advice is say not to statins and avoid the real cause of heart disease……refined carbs and omega six fats.
Jack,
I'm a vegeterian – I don't (and won't) eat meat or fish, but do eat eggs and dairy products. What do you recommend I do?
I've no doubt I have a leptin problem as my BMI is 42, and I also have PCOS.
By the way, my Insulin and Corisol levels (recently tested) are still normal, fortunately, but my CRP level is very high – 22.40.
Thanks
D I am sorry to tell you if you eat that way your future will be compromised from a health stand point. I think you need to strongly reconsider your position. You are eating an incongruent diet to our biologic make up and biochemistry. There is no fix for that mismatch. Good Luck
Is there no way to improve my eating to some extent?
Not if you remain a vegetarian I'm afraid. I would strongly recommend you read Robb Wolf's book, The Paleo Solution. It explains why you need to change
This was very informative. I have been reading your blog alot over the past few days and now its in my bookmarks.
I truly appreciate this blog article.Really thank you! Awesome.
It's hard to seek out educated individuals on this subject, but you sound like you know what you're talking about! Thanks
Here is the link to Dr. Diamond's talk. It is long, If you want to get to the heart of his talk, start at about the 40 minute mark.
http://www.youtube.com/watch?v=3vr-c8GeT34
@D, dude please rethink your ethics in light of what your metabolism is screaming at you to do. What you are doing is not healthy. You will not get healthy until you change your paradigm.
Im obliged for the article post. Really thank you! Want more.
Thank you for this great post! It has been extremely helpful. I hope that you'll proceed posting your wisdom with us.
Dr. Kruse, in some future post, would you be open to listing the tests that you include in your QSelf Assessment each quarter? Including what you think are the resulting normal ranges would also be greatly appreciated.
I may do that in the future.
For more reading I would suggest getting your hands on this article as well by J Abramson et al. "Are lipid lowering guidelines evidence based?," The Lancet, no 369 (2007):168-69. This was an editorial on heart disease prevention trials (8 total) that reveal statins were completely ineffective in reducing overall death risk from heart disease. The study found that the risk of CV events was only minimally reduced by statins. The data revealed that 67 people would need to be treated for five years for just one medical event to be prevented. That event was a heart attack. The trials showed that statins did nothing to lower the death rates at all. The biggest shocker of the trial is this one……….STATINS SHOWED NO BENEFIT IN WOMEN AT ANY AGE! I wonder how many women even know this?
Great info! I love to see someone openly questioning conventional medical "wisdom." It is so compromised by the pharmaceutical co. influence and also by not seeing a problem in a different way. It might be helpful for the general public to understand that drugs are tested and considered to be effective when 50% +1 of the tested population finds effective results. The other 49% percent may show no effect or may have adverse reactions. So our "one size fits all" mentality gives us the current less than perfect system. Thanks so much!
@D – I suffered PCOS on an almost meatless diet. Low carb with a majority of my calories coming from meat was the only cure (and now you could set a clock by my reasonably painless periods.)
If you of the unwilling to switch from vegetarian are concerns about mortality, here are 2 alternative thoughts to chew on:
1)Plants are also life. I have no idea why vegetarians ignore this fact. There is no escaping killing life forms to remain alive. The nature of life is always intertwined with death, as much as we wish otherwise. 2)As far as I can tell the idea that vegetarianism is more "moral" comes to us from India. Hindu society is highly hierarchical, with all the animals, plants, and people in their neat, unchangeable slots. I fail to see how much mortality comes from the idea that some life forms and human lives are more important and better than others. We can do without untouchables and literal sacred cows.
For those of you who still think cholesterol cause heart disease……take a peak at this link. http://www.perfecthealthdiet.com/?p=3836
http://www.karendecoster.com/a-cardiac-surgeo-on-the-glo...
heart disease is a misnomer; the underlying disease process reduces the supply of blood to the heart and other organs leading to angina heart cramp, heart attack and stroke. The disease is characterized by scab-like build-ups that grow on the walls of blood vessels. The correct terminology for this disease process is chronic scurvy, a slower form of the classic vitamin C deficiency disease. collagen supplements The hypothesis that CVD is an ascorbic acid vitamin C deficiency disease was first conceived and tested in the early 1950s Willis devised a method of photographing plaques with X-rays and observed a strange phenomenon in his heart patients. Willis saw that atherosclerotic plaques were not uniformly distributed throughout the vascular system; rather these "blockages" are concentrated near the heart, where arteries are constantly bent or squeezed. Another Canadian, Paterson, had found that the tissues of heart patients were generally depleted of ascorbate vitamin C and it was well known that vitamin C is required for strong and healthy arteries. Willis reasoned that only the mechanical stress caused by the pulse could explain the typical pattern of atherosclerosis that he so often observed in different patients. To Willis, the body was laying down plaque precisely where it was needed in order to stabilize the vascular system. By the late 1980s, medical researchers had made several intriguing discoveries. First came the discovery that heart disease begins with a lesion, a crack or stress fracture, in the arterial wall. The question became, and remains, as to the cause of these lesions in human beings since they do not arise in most other animals. Then a variant of the so-called "bad" LDL cholesterol called lipoprotein(a), or Lp(a) for short, was studied and found to be really bad. It is sticky because of receptors on the surface of the molecule called lysine binding sites. Work that led to the 1987 Nobel prize in medicine discovered that lysine and proline binding sites cause the formation of atherosclerotic plaques. Then, Beisiegel et. al. in Germany examined plaques post mortem and found only Lp(a), not ordinary LDL cholesterol.
I've been exploring for a little for any high-quality articles or weblog posts on this kind of area . Exploring in Yahoo I ultimately stumbled upon this web site. Studying this info So I'm satisfied to convey that I've a very excellent uncanny feeling I discovered exactly what I needed. I most without a doubt will make certain to do not overlook this web site and provides it a glance regularly.
http://drrosedale.com/resources/pdf/Cholesterol%2…
Nice article here for folks to read.
This video is a MUST SEE. It confirms the Paleo Diet, and proves that the Standard American Diet is totally unhealthy & wrong because of corruption & lies all for profit at the expense of American Health.
How Bad Science and Big Business Created the Obesity Epidemic
http://www.youtube.com/watch?v=3vr-c8GeT34
This video should be posted on every facebook page, and sent out to everyone's email list.
Many thanks to Dr. Kruse for bringing this exceptional Expose' to our attention. This has to be the most important video of our time, well worth watching!
Grizz
I see much mentioned about Heart disease, cholestrerol and statins but not much mentioned about high blood pressure and how to treat it. I have hypothyroidism and high blood pressure but my cholsterol is fine. Whats up?
@Les if you eat a paleo diet and use my leptin Rx you wont have to worry about high blood pressure long term…..it demolishes it.
Well, the NIH has finally said it! It's not sat fats or cholesterol that cause cardiovascular disease–it's the combination of sat fats WITH HIGH CARB INTAKE, PARTICULARLY those with a HIGH GLYCEMIC INDEX. (Yes, I'm shouting the news!) Low-carbers have been saying it for years, Dr Atkins having led the charge.
http://www.ncbi.nlm.nih.gov/pubmed/21978979
@Mimi Did we really need them to tell us this? I think most of us in the primal/paleo world get this implicitly.
I know, I'm just so excited that they're getting it!! 🙂 A doctor I know took the link down so she can share it to defend the low-carb diet with a conventional-medicine source! Great way to spread the re-education. 🙂 Those us on LC get tired of the misinformation, don't we…nice to be able to throw a CW source at it. 🙂
@harry If your doc tells you he reads about heart disease show him this link listed below.
For instance, a recent study published in the 2009 Journal of American Medical Association studied the evidence supporting the practice guidelines and consensus statements published by the American College of Cardiology and the American Heart Association. It was found that only 11% of the recommendations, practice guidelines and consensus statements were based on quality evidence and over half were based on poor quality evidence that was little more than the panel's opinion. The review also found that even the strongest (Class 1) recommendations, which are considered medical dogma, cited as a legal standards and often go unquestioned as medical fact, were only supported by high quality evidence 19% of the time and not revised based on new evidence.
1.Tricoci P, Allen JM, Kramer KM, et al. Scientific evidnce underlying the ACC/AHA clincal practice guidelines. JAMA 2009;301(8):831-841.
Slightly more people die on statin treatment
This study was published in the Lancet 2008 Oct 4;372(9645):1231-9
Everyone should read this one!!!
http://drhyman.com/do-statins-cause-diabetes-and-…
I think there must be a confusing typo in this line in paragraph 2 that should be fixed: "Framingham data was REPORTED to say that heart disease correlated with heart disease best." It should say "heart disease correlated with high cholesterol levels best" or something along those lines?
I am 57 yrs old with a brain disease. NOthing in my body worked right. I had a TIA last week and probably have been having them for several yrs. My cholesterol is 164 and I have HBP now. My doctor put me on Plavix, aspirin and Atorvastin. I don't want to be on the Plavix or Stain in particular. I just started Paleo. Will my cholesterol go down and will the plaque build up go away? I don't know what to do. My case is complicated because I have Cerebellar Atrophy secondary to radiation tx as an infant.
@Gloria The XRT as a child to your posterior fossa in your brain worries me. That means the major nerve that covers your gut is likely not working well and you have a leaky gut because of it. You need to find a functional medicine doctor close to you to help you. Go google funtionalmedicine.org to find one near you
Oops! My cholesterol is 264 not 164. I used to have low blood pressure but now since my TIA.
http://health.msn.com/health-topics/cancer/low-bad-cholesterol-levels-may-be-linked-to-cancer-risk
http://www.dietheartpublishing.com/diet-heart-timeline
I sent this blog post to a Doctor/Teacher friend of mine.
This is his response:
Dear Doug,
The lipid hypothesis is alive and well, but it is evolving in ways that are difficult to capture in simplistic blogs like the one you sent me. Lipid particle size and subclasses of each lipoprotein are vitally important in ways that we are just beginning to understand. And most cardiologists today realize that lipoprotein particles are just part of the story.
As for the paleolithic diet there is a lot to like about it. But any time that you eliminate a whole food class (in this case whole grains) you end up with an unbalanced diet. My preference is the Mediterranean diet with a bit more emphasis on low fat protein sources.
@Douglas…..if he were my doc I’d fire him…….whole grains are not balanced for anyone. He needs to read Cordain’s work more. Buy him The Paleo Answer by Cordain and tell him to really read the bibliography about grains……then get him Wheat Belly written by a cardiologist William Davis. Then tell him how simplistic my blog is………..
http://dietheartnews.com/2012/08/illustrated-history-of-heart-disease-1825-2015/
Oh yes, the wonders of Lipitor. I have a brother-in-law who took it and sung it’s praises. He has been in a home for Alzheimers patients for several years. And, he is a MD. I am an RN and tried talking to him about associated memory loss and he ignored me.
Love what you are doing!
Thanks Casey